ID 41176 Auteurs Hélène Touzet Mikaël Salson Claire Lemaitre Image puzzle-bioinfo Domaines applicatifs Santé - Médecine personnalisée Comment la bioinformatique a résolu le puzzle du génome du SARS-CoV-2 Contenu Séquençage du matériel génétique L'identification de l'agent causal d'une infection inconnue passe généralement par le séquençage de son matériel génétique, c'est-à -dire la détermination de la séquence des nucléotides (les A, T, C et G) qui composent son génome. Depuis le milieu des années 2010, le séquençage à haut débit couplé à la bio-informatique permet de caractériser le génome d'un virus émergent en quelques jours pour quelques centaines d'euros. Ainsi, de décembre 2019 à janvier 2020, plusieurs hôpitaux de Wuhan en Chine confrontés à la maladie se sont lancés indépendamment dans le séquençage du nouvel agent pathogène, en suivant approximativement le même protocole. Le point de départ consiste à collecter le fluide pulmonaire de patients, puis à en extraire le matériel génétique. L’ADN ainsi obtenu est prêt à être séquencé. Lors du séquençage, les molécules d’ADN ou d’ARN ne peuvent pas être obtenues de bout en bout car la technologie utilisée ne produit que de courts fragments de séquences d’environ 200 nucléotides chacun. À l’issue du séquençage, les données brutes sont ainsi une soupe de centaines de millions de courtes séquences nucléiques, appelées lectures, qui couvrent de manière aléatoire les génomes initiaux. Reconstituer le génome d'intérêt nécessite ensuite toute une série de traitements informatiques. Identification des lectures d’intérêt Le premier problème est que les données de séquençage sont mélangées. Elles proviennent de tous les micro-organismes présents dans l'échantillon clinique possiblement associés aux cellules humaines environnantes. Il faut donc faire le tri dans ce microbiome pulmonaire. Cela est réalisé par la fouille de grandes bases de données génomiques comprenant les génomes de l’ensemble des microbes connus (virus, bactéries, champignons et parasites) ainsi que le génome humain. Pour cela, la communauté bio-informatique a développé dès la fin des années 1990 des moteurs de recherche génomique, tels que Blast, capables de traiter efficacement des grandes masses de séquences à l’image d’un Google pour ADN. Ces outils calculent des alignements de séquences qui identifient les lectures similaires aux séquences présentes dans la base de données et distinguent ainsi les lectures provenant potentiellement du nouveau virus (voir la figure 1 ci-dessous, à droite). Les méthodes les plus récentes ont été conçues depuis 2009-2010 pour traiter spécifiquement les lectures de séquençage à haut débit. Le cÅ“ur algorithmique s'appuie sur des concepts avancés issus de la théorie de l'information, tels que la compression, les fonctions de hachage, les structures de données d'indexation. On peut ainsi en quelques minutes de calcul isoler la fraction des lectures provenant du nouveau virus, fraction qui représente généralement moins de 1 % des données initiales.  [caption id="attachment_44292" align="aligncenter" width="650"] Figure 1 : Alignement de séquences biologiques.[/caption] Contenu Séquençage du matériel génétique L'identification de l'agent causal d'une infection inconnue passe généralement par le séquençage de son matériel génétique, c'est-à -dire la détermination de la séquence des nucléotides (les A, T, C et G) qui composent son génome. Depuis le milieu des années 2010, le séquençage à haut débit couplé à la bio-informatique permet de caractériser le génome d'un virus émergent en quelques jours pour quelques centaines d'euros. Ainsi, de décembre 2019 à janvier 2020, plusieurs hôpitaux de Wuhan en Chine confrontés à la maladie se sont lancés indépendamment dans le séquençage du nouvel agent pathogène, en suivant approximativement le même protocole. Le point de départ consiste à collecter le fluide pulmonaire de patients, puis à en extraire le matériel génétique. L’ADN ainsi obtenu est prêt à être séquencé. Lors du séquençage, les molécules d’ADN ou d’ARN ne peuvent pas être obtenues de bout en bout car la technologie utilisée ne produit que de courts fragments de séquences d’environ 200 nucléotides chacun. À l’issue du séquençage, les données brutes sont ainsi une soupe de centaines de millions de courtes séquences nucléiques, appelées lectures, qui couvrent de manière aléatoire les génomes initiaux. Reconstituer le génome d'intérêt nécessite ensuite toute une série de traitements informatiques. Identification des lectures d’intérêt Le premier problème est que les données de séquençage sont mélangées. Elles proviennent de tous les micro-organismes présents dans l'échantillon clinique possiblement associés aux cellules humaines environnantes. Il faut donc faire le tri dans ce microbiome pulmonaire. Cela est réalisé par la fouille de grandes bases de données génomiques comprenant les génomes de l’ensemble des microbes connus (virus, bactéries, champignons et parasites) ainsi que le génome humain. Pour cela, la communauté bio-informatique a développé dès la fin des années 1990 des moteurs de recherche génomique, tels que Blast, capables de traiter efficacement des grandes masses de séquences à l’image d’un Google pour ADN. Ces outils calculent des alignements de séquences qui identifient les lectures similaires aux séquences présentes dans la base de données et distinguent ainsi les lectures provenant potentiellement du nouveau virus (voir la figure 1 ci-dessous, à droite). Les méthodes les plus récentes ont été conçues depuis 2009-2010 pour traiter spécifiquement les lectures de séquençage à haut débit. Le cÅ“ur algorithmique s'appuie sur des concepts avancés issus de la théorie de l'information, tels que la compression, les fonctions de hachage, les structures de données d'indexation. On peut ainsi en quelques minutes de calcul isoler la fraction des lectures provenant du nouveau virus, fraction qui représente généralement moins de 1 % des données initiales.  [caption id="attachment_44292" align="aligncenter" width="650"] Figure 1 : Alignement de séquences biologiques.[/caption]
ID 41176 Auteurs Hélène Touzet Mikaël Salson Claire Lemaitre Contenu Séquençage du matériel génétique L'identification de l'agent causal d'une infection inconnue passe généralement par le séquençage de son matériel génétique, c'est-à -dire la détermination de la séquence des nucléotides (les A, T, C et G) qui composent son génome. Depuis le milieu des années 2010, le séquençage à haut débit couplé à la bio-informatique permet de caractériser le génome d'un virus émergent en quelques jours pour quelques centaines d'euros. Ainsi, de décembre 2019 à janvier 2020, plusieurs hôpitaux de Wuhan en Chine confrontés à la maladie se sont lancés indépendamment dans le séquençage du nouvel agent pathogène, en suivant approximativement le même protocole. Le point de départ consiste à collecter le fluide pulmonaire de patients, puis à en extraire le matériel génétique. L’ADN ainsi obtenu est prêt à être séquencé. Lors du séquençage, les molécules d’ADN ou d’ARN ne peuvent pas être obtenues de bout en bout car la technologie utilisée ne produit que de courts fragments de séquences d’environ 200 nucléotides chacun. À l’issue du séquençage, les données brutes sont ainsi une soupe de centaines de millions de courtes séquences nucléiques, appelées lectures, qui couvrent de manière aléatoire les génomes initiaux. Reconstituer le génome d'intérêt nécessite ensuite toute une série de traitements informatiques. Identification des lectures d’intérêt Le premier problème est que les données de séquençage sont mélangées. Elles proviennent de tous les micro-organismes présents dans l'échantillon clinique possiblement associés aux cellules humaines environnantes. Il faut donc faire le tri dans ce microbiome pulmonaire. Cela est réalisé par la fouille de grandes bases de données génomiques comprenant les génomes de l’ensemble des microbes connus (virus, bactéries, champignons et parasites) ainsi que le génome humain. Pour cela, la communauté bio-informatique a développé dès la fin des années 1990 des moteurs de recherche génomique, tels que Blast, capables de traiter efficacement des grandes masses de séquences à l’image d’un Google pour ADN. Ces outils calculent des alignements de séquences qui identifient les lectures similaires aux séquences présentes dans la base de données et distinguent ainsi les lectures provenant potentiellement du nouveau virus (voir la figure 1 ci-dessous, à droite). Les méthodes les plus récentes ont été conçues depuis 2009-2010 pour traiter spécifiquement les lectures de séquençage à haut débit. Le cÅ“ur algorithmique s'appuie sur des concepts avancés issus de la théorie de l'information, tels que la compression, les fonctions de hachage, les structures de données d'indexation. On peut ainsi en quelques minutes de calcul isoler la fraction des lectures provenant du nouveau virus, fraction qui représente généralement moins de 1 % des données initiales.  [caption id="attachment_44292" align="aligncenter" width="650"] Figure 1 : Alignement de séquences biologiques.[/caption] Image puzzle-bioinfo Domaines applicatifs Santé - Médecine personnalisée
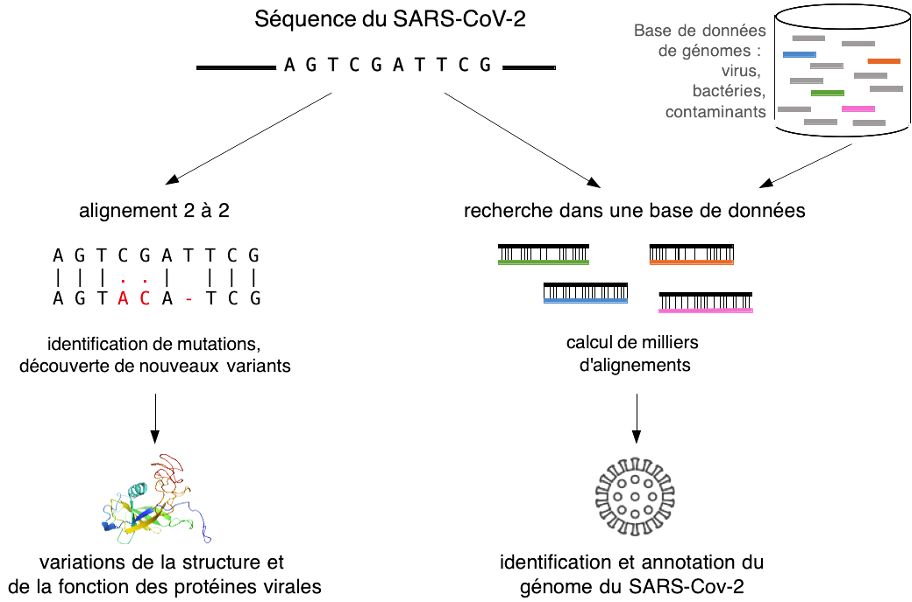 Figure 1 : Alignement de séquences biologiques.[/caption]
Figure 1 : Alignement de séquences biologiques.[/caption]