Le SARS-CoV-2 : une expérience inédite de surveillance génomique mondiale
En effet, le génome d’un virus est par nature très dynamique, avec une évolution qui se manifeste par l’accumulation rapide de mutations. Disposer au fil du temps de nombreux génomes d’origines géographiques variées est donc nécessaire pour identifier l’émergence de variants, des lignées porteuses de mutations clés susceptibles d’affecter la pathogénicité et la transmissibilité du virus, voire de mener à un échappement vaccinal. Ce type de surveillance a pu être expérimenté ces dernières années avec la grippe saisonnière, les virus Ebola ou Zika, et a atteint une ampleur inédite avec le suivi du SARS-CoV-2. Une telle tâche requiert des moyens de génération, d’analyse bio-informatique et de partage des données particulièrement optimisés et ambitieux. Comment cela se passe-t-il ?
Séquençage à grande échelle
En janvier 2020, la découverte des premiers génomes du SARS-CoV-2 à Wuhan avait nécessité des techniques de séquençage et d’analyse bio-informatique assez complexes (voir notre article « Comment la bio-informatique a résolu le puzzle du génome du SARS-CoV-2 »). Il avait fallu réaliser le séquençage de l'ensemble du contenu en ARN d’échantillons pulmonaires des patients, même si la fraction virale contenue dans ces prélèvements est extrêmement faible. Ce type de séquençage produit de grandes quantités de données de séquences et nécessite ensuite la mise en œuvre d’analyses bio-informatiques sophistiquées et coûteuses. Tout cela est peu compatible avec un suivi routinier de l’épidémie. Aujourd’hui, maintenant que le génome de référence est connu, il est possible à la fois de réduire les coûts de séquençage en ciblant exclusivement les séquences d'intérêt désormais connues dans l’échantillon, et d’accélérer les traitements bio-informatiques d’assemblage et d’analyse des nouveaux génomes.
Dans le cas du SARS-CoV-2, la stratégie la plus utilisée pour cibler le génome viral est le séquençage par amplicons. Dans ce protocole, le matériel génétique du virus présent dans l’échantillon biologique est d’abord amplifié par PCR (réaction en chaîne par polymérase), puis les fragments d’ADN issus de l’amplification (les amplicons) sont séquencés. La PCR est une technique moléculaire largement utilisée en biologie qui repose notamment sur une première étape bio-informatique cruciale : la conception d’amorces, des petites séquences bien choisies sur le génome cible (voir Encadré 1).
Une fois tous les amplicons séquencés, l'assemblage du génome viral est également facilité par l'utilisation de la séquence génomique déjà connue de SARS-CoV-2, qui sert de modèle. L'assemblage de novo (voir « Comment la bioinformatique a résolu le puzzle du génome du SARS-CoV-2 » pour plus de détails) n'est plus nécessaire, et les lectures sont simplement alignées sur le génome de référence. L'alignement d'une lecture sur le génome de référence consiste à la positionner sur ce génome, c'est-à -dire identifier la portion du génome qui présente le plus de similarité avec la lecture, et à lister les caractères identiques et différents entre ces deux séquences. Une fois toutes les lectures alignées, une séquence consensus du génome peut alors être calculée en sélectionnant, à chaque position, le caractère observé dans la majorité des lectures alignées à la position donnée. Les variations de séquences, telles que les mutations, sont ensuite identifiées entre la souche virale nouvellement séquencée et le génome de référence ou entre différentes souches coexistant dans l'échantillon. La Figure 1 ci-dessous montre les mutations qui ont été identifiées dans le gène de la protéine S dans différentes lignées du SARS-CoV-2.
[caption id="attachment_48126" align="aligncenter" width="750"]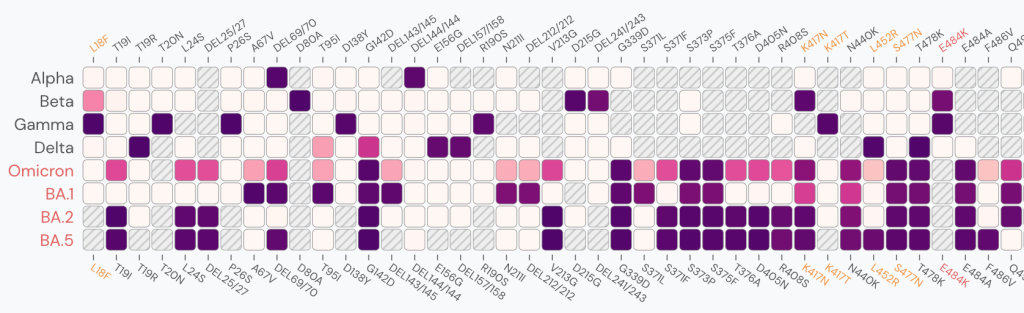 Figure 1 : Prévalence des mutations identifiées dans le gène de la protéine S (positions sur le gène en abscisse) dans les différentes lignées de SARS-CoV-2 (en ordonnée, BA.1, BA.2 et BA.5 sont des sous-variants d’Omicron). Le dégradé de couleur indique la proportion des génomes de la lignée dans lesquels la mutation a été observée (une case est grisée quand la mutation n’a encore jamais été détectée dans la lignée ; la couleur blanche indique que la mutation a été observée dans très peu de génomes de la lignée, la couleur violet foncé indique que la mutation a été observée dans 100 % des génomes de la lignée). Figure générée par le site outbreak.info qui utilise les données de la base de données GISAID.[/caption]
Figure 1 : Prévalence des mutations identifiées dans le gène de la protéine S (positions sur le gène en abscisse) dans les différentes lignées de SARS-CoV-2 (en ordonnée, BA.1, BA.2 et BA.5 sont des sous-variants d’Omicron). Le dégradé de couleur indique la proportion des génomes de la lignée dans lesquels la mutation a été observée (une case est grisée quand la mutation n’a encore jamais été détectée dans la lignée ; la couleur blanche indique que la mutation a été observée dans très peu de génomes de la lignée, la couleur violet foncé indique que la mutation a été observée dans 100 % des génomes de la lignée). Figure générée par le site outbreak.info qui utilise les données de la base de données GISAID.[/caption]
L’organisation et le partage des informations
En juin 2021, un an et demi après la publication du premier génome de SARS-CoV-2 repéré à Wuhan, plus de 2 millions de génomes de SARS-CoV-2 ont été séquencés, assemblés et partagés par divers laboratoires et institutions de pays du monde entier. Ce vaste effort de séquençage permet de comprendre comment le virus évolue, de suivre les mutations en temps réel et d'identifier de nouveaux variants. Un aspect important de cette recherche est que plusieurs initiatives nationales et internationales ont rapidement développé des portails web dédiés afin de stocker ces informations et de les rendre librement disponibles sur Internet. Les dépôts de séquences généralistes tels que Genbank, hébergé par le NCBI (États-Unis), ou l'ENA (European Nucleotide Archive), hébergé par l'European Bioinformatics Institute, qui organisent le partage des données de séquences du domaine public depuis plusieurs décennies, ont développé des bases de données et des outils spécifiques pour les données du SARS-CoV-2. Le Consortium GISAID fournit également une ressource essentielle pour les génomes de SARS-CoV-2 (ressource disponible sur inscription). La base de données GISAID comptait 339 génomes du SARS-CoV-2 disponibles à la fin du mois de janvier 2020, et ce nombre a augmenté rapidement, atteignant environ 80 000 en août 2020, 1 million début avril 2021, puis plus de 4 millions six mois plus tard et plus de 14 millions en novembre 2022 (voir Figure 2). Lorsque l’activité épidémique est importante, 1 million de séquences peuvent être ajoutées en un seul mois.
Ce sont les scientifiques qui soumettent librement des données de séquences à de telles collections. Ces séquences sont vérifiées avant d'être partagées publiquement. Les séquences génomiques disponibles sont également parfois accompagnées d'informations supplémentaires, telles que l'origine géographique et la date de collecte de l'échantillon, les protocoles de séquençage, les informations cliniques du patient, etc. Ces métadonnées sont structurées dans des bases de données pour permettre des requêtes efficaces et des analyses comparatives en aval à partir de cette énorme collection de séquences.
[caption id="attachment_48127" align="aligncenter" width="748"]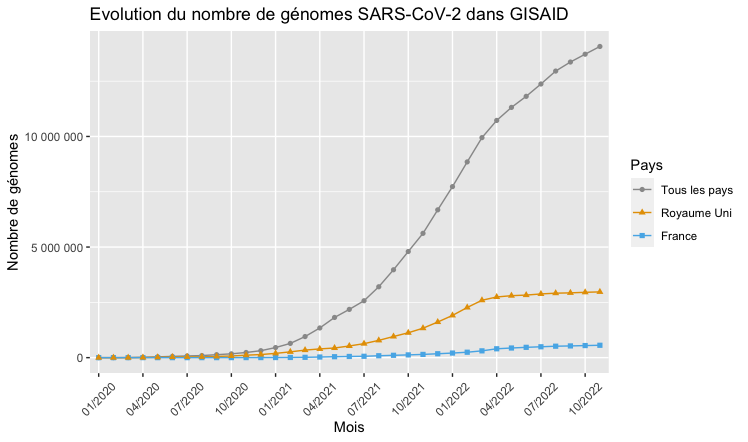 Figure 2 : Évolution du nombre de génomes de SARS-CoV-2 déposés dans la base de données GISAID entre janvier 2020 et novembre 2022 au niveau mondial (en gris), par le Royaume-Uni (en jaune) et par la France (en bleu). Figure produite à partir des statistiques fournies par GISAID.[/caption]
Figure 2 : Évolution du nombre de génomes de SARS-CoV-2 déposés dans la base de données GISAID entre janvier 2020 et novembre 2022 au niveau mondial (en gris), par le Royaume-Uni (en jaune) et par la France (en bleu). Figure produite à partir des statistiques fournies par GISAID.[/caption]
Toutes ces informations (génomes et métadonnées) sont utilisées pour suivre la diversité de la population de SARS-CoV-2 dans le monde. Mais ces données servent également à détecter de manière précoce des variants préoccupants. Alerté par la reprise du nombre d’infections dans le Sud-Est de l’Angleterre à l’automne 2020, le Royaume-Uni a pu expliquer ce phénomène par la propagation d’un nouveau variant, de la lignée B.1.1.7, et suivre la propagation de ce qui a ensuite été appelé le variant Alpha. En étudiant rétrospectivement les données de séquençage, on a pu constater que ce variant avait été initialement détecté en septembre 2020. De même, à la fin du printemps 2021, le Royaume-Uni a pu associer une nouvelle reprise épidémique sur son territoire à la propagation du variant Delta, initialement détecté en Inde. De manière similaire, l’Afrique du Sud, qui dispose de moyens importants de séquençage et d’analyse bio-informatique pour le suivi de l’épidémie, a détecté le variant Omicron à l’automne 2021 (voir par exemple l'article en anglais « African coronavirus surveillance network provides early warning for world »). Ces découvertes ont été facilitées par une politique volontariste de surveillance génomique, que l’Organisation mondiale de la Santé (OMS) recommande afin de détecter au plus tôt l’émergence de variants préoccupants. À ce titre, le Royaume-Uni figure parmi les pionniers. En 2020, plus de 140 000 génomes avaient été publiés via le consortium COG-UK, contre moins de 3 000 en France, qui n’avait alors pas encore de consortium en place. Un tel consortium a depuis été mis en place en France à partir du début de l’année 2021 et a publié plus de 170 000 séquences cette année-là . L’Institut Français de Bio-informatique (IFB) apporte un soutien technique avec notamment la mise en place et la gestion d’une base de données sécurisée. Le Royaume-Uni a publié 1,5 million de séquences en 2021 (voir Figure 2).
En l’état actuel des connaissances, un génome n’est cependant toujours pas suffisant pour caractériser un phénotype, et il reste indispensable de coupler les jeux de séquences génomiques avec d’autres types de données (nombres de cas, statut vaccinal des patients, informations sur la sévérité de l’infection, une éventuelle hospitalisation, etc.). Toutes ces questions sont au cœur de l’épidémiologie génomique, discipline récente rendue possible par les progrès du séquençage et de la bio-informatique. La pandémie de SARS-CoV-2 montre toute l’importance d’une politique de science ouverte ambitieuse et volontaire, avec le stockage, la structuration et le partage des données.
En effet, le génome d’un virus est par nature très dynamique, avec une évolution qui se manifeste par l’accumulation rapide de mutations. Disposer au fil du temps de nombreux génomes d’origines géographiques variées est donc nécessaire pour identifier l’émergence de variants, des lignées porteuses de mutations clés susceptibles d’affecter la pathogénicité et la transmissibilité du virus, voire de mener à un échappement vaccinal. Ce type de surveillance a pu être expérimenté ces dernières années avec la grippe saisonnière, les virus Ebola ou Zika, et a atteint une ampleur inédite avec le suivi du SARS-CoV-2. Une telle tâche requiert des moyens de génération, d’analyse bio-informatique et de partage des données particulièrement optimisés et ambitieux. Comment cela se passe-t-il ?
Séquençage à grande échelle
En janvier 2020, la découverte des premiers génomes du SARS-CoV-2 à Wuhan avait nécessité des techniques de séquençage et d’analyse bio-informatique assez complexes (voir notre article « Comment la bio-informatique a résolu le puzzle du génome du SARS-CoV-2 »). Il avait fallu réaliser le séquençage de l'ensemble du contenu en ARN d’échantillons pulmonaires des patients, même si la fraction virale contenue dans ces prélèvements est extrêmement faible. Ce type de séquençage produit de grandes quantités de données de séquences et nécessite ensuite la mise en œuvre d’analyses bio-informatiques sophistiquées et coûteuses. Tout cela est peu compatible avec un suivi routinier de l’épidémie. Aujourd’hui, maintenant que le génome de référence est connu, il est possible à la fois de réduire les coûts de séquençage en ciblant exclusivement les séquences d'intérêt désormais connues dans l’échantillon, et d’accélérer les traitements bio-informatiques d’assemblage et d’analyse des nouveaux génomes.
Dans le cas du SARS-CoV-2, la stratégie la plus utilisée pour cibler le génome viral est le séquençage par amplicons. Dans ce protocole, le matériel génétique du virus présent dans l’échantillon biologique est d’abord amplifié par PCR (réaction en chaîne par polymérase), puis les fragments d’ADN issus de l’amplification (les amplicons) sont séquencés. La PCR est une technique moléculaire largement utilisée en biologie qui repose notamment sur une première étape bio-informatique cruciale : la conception d’amorces, des petites séquences bien choisies sur le génome cible (voir Encadré 1).
Une fois tous les amplicons séquencés, l'assemblage du génome viral est également facilité par l'utilisation de la séquence génomique déjà connue de SARS-CoV-2, qui sert de modèle. L'assemblage de novo (voir « Comment la bioinformatique a résolu le puzzle du génome du SARS-CoV-2 » pour plus de détails) n'est plus nécessaire, et les lectures sont simplement alignées sur le génome de référence. L'alignement d'une lecture sur le génome de référence consiste à la positionner sur ce génome, c'est-à -dire identifier la portion du génome qui présente le plus de similarité avec la lecture, et à lister les caractères identiques et différents entre ces deux séquences. Une fois toutes les lectures alignées, une séquence consensus du génome peut alors être calculée en sélectionnant, à chaque position, le caractère observé dans la majorité des lectures alignées à la position donnée. Les variations de séquences, telles que les mutations, sont ensuite identifiées entre la souche virale nouvellement séquencée et le génome de référence ou entre différentes souches coexistant dans l'échantillon. La Figure 1 ci-dessous montre les mutations qui ont été identifiées dans le gène de la protéine S dans différentes lignées du SARS-CoV-2.
[caption id="attachment_48126" align="aligncenter" width="750"] Figure 1 : Prévalence des mutations identifiées dans le gène de la protéine S (positions sur le gène en abscisse) dans les différentes lignées de SARS-CoV-2 (en ordonnée, BA.1, BA.2 et BA.5 sont des sous-variants d’Omicron). Le dégradé de couleur indique la proportion des génomes de la lignée dans lesquels la mutation a été observée (une case est grisée quand la mutation n’a encore jamais été détectée dans la lignée ; la couleur blanche indique que la mutation a été observée dans très peu de génomes de la lignée, la couleur violet foncé indique que la mutation a été observée dans 100 % des génomes de la lignée). Figure générée par le site outbreak.info qui utilise les données de la base de données GISAID.[/caption]
L’organisation et le partage des informations
En juin 2021, un an et demi après la publication du premier génome de SARS-CoV-2 repéré à Wuhan, plus de 2 millions de génomes de SARS-CoV-2 ont été séquencés, assemblés et partagés par divers laboratoires et institutions de pays du monde entier. Ce vaste effort de séquençage permet de comprendre comment le virus évolue, de suivre les mutations en temps réel et d'identifier de nouveaux variants. Un aspect important de cette recherche est que plusieurs initiatives nationales et internationales ont rapidement développé des portails web dédiés afin de stocker ces informations et de les rendre librement disponibles sur Internet. Les dépôts de séquences généralistes tels que Genbank, hébergé par le NCBI (États-Unis), ou l'ENA (European Nucleotide Archive), hébergé par l'European Bioinformatics Institute, qui organisent le partage des données de séquences du domaine public depuis plusieurs décennies, ont développé des bases de données et des outils spécifiques pour les données du SARS-CoV-2. Le Consortium GISAID fournit également une ressource essentielle pour les génomes de SARS-CoV-2 (ressource disponible sur inscription). La base de données GISAID comptait 339 génomes du SARS-CoV-2 disponibles à la fin du mois de janvier 2020, et ce nombre a augmenté rapidement, atteignant environ 80 000 en août 2020, 1 million début avril 2021, puis plus de 4 millions six mois plus tard et plus de 14 millions en novembre 2022 (voir Figure 2). Lorsque l’activité épidémique est importante, 1 million de séquences peuvent être ajoutées en un seul mois.
Ce sont les scientifiques qui soumettent librement des données de séquences à de telles collections. Ces séquences sont vérifiées avant d'être partagées publiquement. Les séquences génomiques disponibles sont également parfois accompagnées d'informations supplémentaires, telles que l'origine géographique et la date de collecte de l'échantillon, les protocoles de séquençage, les informations cliniques du patient, etc. Ces métadonnées sont structurées dans des bases de données pour permettre des requêtes efficaces et des analyses comparatives en aval à partir de cette énorme collection de séquences.
[caption id="attachment_48127" align="aligncenter" width="748"] Figure 2 : Évolution du nombre de génomes de SARS-CoV-2 déposés dans la base de données GISAID entre janvier 2020 et novembre 2022 au niveau mondial (en gris), par le Royaume-Uni (en jaune) et par la France (en bleu). Figure produite à partir des statistiques fournies par GISAID.[/caption]
Toutes ces informations (génomes et métadonnées) sont utilisées pour suivre la diversité de la population de SARS-CoV-2 dans le monde. Mais ces données servent également à détecter de manière précoce des variants préoccupants. Alerté par la reprise du nombre d’infections dans le Sud-Est de l’Angleterre à l’automne 2020, le Royaume-Uni a pu expliquer ce phénomène par la propagation d’un nouveau variant, de la lignée B.1.1.7, et suivre la propagation de ce qui a ensuite été appelé le variant Alpha. En étudiant rétrospectivement les données de séquençage, on a pu constater que ce variant avait été initialement détecté en septembre 2020. De même, à la fin du printemps 2021, le Royaume-Uni a pu associer une nouvelle reprise épidémique sur son territoire à la propagation du variant Delta, initialement détecté en Inde. De manière similaire, l’Afrique du Sud, qui dispose de moyens importants de séquençage et d’analyse bio-informatique pour le suivi de l’épidémie, a détecté le variant Omicron à l’automne 2021 (voir par exemple l'article en anglais « African coronavirus surveillance network provides early warning for world »). Ces découvertes ont été facilitées par une politique volontariste de surveillance génomique, que l’Organisation mondiale de la Santé (OMS) recommande afin de détecter au plus tôt l’émergence de variants préoccupants. À ce titre, le Royaume-Uni figure parmi les pionniers. En 2020, plus de 140 000 génomes avaient été publiés via le consortium COG-UK, contre moins de 3 000 en France, qui n’avait alors pas encore de consortium en place. Un tel consortium a depuis été mis en place en France à partir du début de l’année 2021 et a publié plus de 170 000 séquences cette année-là . L’Institut Français de Bio-informatique (IFB) apporte un soutien technique avec notamment la mise en place et la gestion d’une base de données sécurisée. Le Royaume-Uni a publié 1,5 million de séquences en 2021 (voir Figure 2).
En l’état actuel des connaissances, un génome n’est cependant toujours pas suffisant pour caractériser un phénotype, et il reste indispensable de coupler les jeux de séquences génomiques avec d’autres types de données (nombres de cas, statut vaccinal des patients, informations sur la sévérité de l’infection, une éventuelle hospitalisation, etc.). Toutes ces questions sont au cœur de l’épidémiologie génomique, discipline récente rendue possible par les progrès du séquençage et de la bio-informatique. La pandémie de SARS-CoV-2 montre toute l’importance d’une politique de science ouverte ambitieuse et volontaire, avec le stockage, la structuration et le partage des données.